 |
In the early 1990s New York City
experienced an outbreak of MDR tuberculosis. At the time we were studying DNA
topoisomerases to better understand chromosome structure. We had used quinolones as tools,
and so we were familiar with them. During the outbreak it became clear that the use of
ciprofloxacin quickly led, sometimes within a month, to ciprofloxacin-resistant
tuberculosis (13, 17). At the time it seemed that the problem was to find a way to halt
the development of resistance. From a naive microbiological point of view we thought that
we should look for new quinolones that would readily attack resistant mutants. We had
gyrase mutants available for several bacterial species, and we obtained some
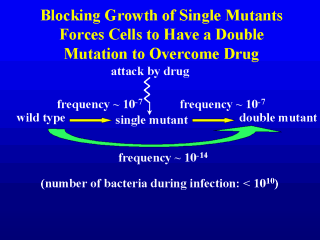
investigational compounds from John Domagala at Parke-Davis. The general idea, as shown in
the slide, was to find a compound that would be exceptionally active against a resistant
mutant. Such a compound, if used against wild-type cells, would require a double mutation
for growth. Since the mutation frequency for fluoroquinolones is on the order of 10-7,
a pair of independent mutations would arise at frequency of 10-14. Infections
rarely have more than 1010 cells, so a mutant-active compound would prevent the
selective enrichment of mutants. The story that emerged can be divided into several
subtopics, as shown on the next slide. |