| front |1 |2 |3 |4 |5 |6 |7 |8 |9 |10 |11 |12 |13 |14 |15 |16 |17 |18 |19 |20 |21 |22 |23 |24 |25 |26 |27 |28 |29 |30 |31 |32 |33 |34 |35 |36 |review |
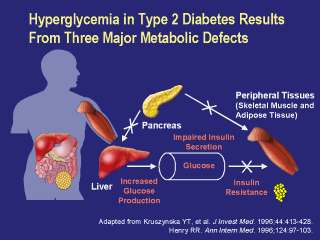 |
Three major metabolic defects contribute to
hyperglycemia in patients with type 2 diabetes: increased hepatic glucose
production, impaired pancreatic insulin secretion, and peripheral tissue
insulin resistance. After eating a meal or ingesting glucose, insulin is secreted, hepatic glucose output is suppressed, and insulin-dependent glucose uptake by peripheral tissues is stimulated. In type 2 diabetes, insulin resistance and impaired insulin secretion inhibit normal suppression of hepatic glucose output. As a consequence, the liver continues to release glucose into the circulation. Moreover, peripheral insulin resistance coupled with insufficient insulin results in decreased uptake of glucose by insulin-dependent target tissues, notably skeletal muscle and adipose tissue. These mechanisms contribute to postprandial hyperglycemia in type 2 diabetes. In type 2 diabetes, increased hepatic glucose production is the primary factor responsible for the fasting hyperglycemia. Moreover, in patients with type 2 diabetes, fasting blood glucose levels correlate strongly with rates of hepatic glucose output. In the setting of peripheral insulin resistance, insulin-mediated glucose uptake cannot accommodate the increased hepatic glucose output and rise in fasting glucose levels. Kruszynska YT, et al. J Invest Med. 1996;44:413-428. Henry RR. Ann Intern Med. 1996;124:97-103. |