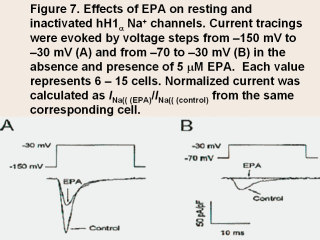
 |
This figure shows the effect of EPA on the
resting myocyte held at a potential of –150 mV (Fig. 7A) and an inactivated
partially depolarized myocytes held at a potential of –70 mV (Fig. 7B) in
hH1α (human myocardial Na+ channels
transiently expressing both the α- and
β1-subunits in HEK293 cells). It can be
seen that from a membrane potential held at –150 mV, even in the presence of
5 µM EPA, the inward Na+ current is
decreased but is still a sufficiently robust INa to induce an action
potential, which would propagate through the heart and cause a normal
cardiac contraction. By contrast, in the partially depolarized cell with a
membrane potential held at –70 mV even the control current was much reduced.
This current, however, would likely induce an aberrant action potential and
with the nonhomogeneous conduction rates of action potentials in the
ischemic myocardium cause a fatal, reentrant arrhythmia. But in the presence
of the same 5 µM concentration of EPA
any INa would be eliminated. This is what we mean in saying that partially
depolarized myocytes would be eliminated from any proarrhythmic effects in
the presence of n-3 PUFAs.
This effect of the n-3 PUFAs on Na+ channels, together with their effect to
inhibit L-type Ca2+ channels preventing triggered arrhythmic after-potential
discharges due to excessive cytosolic Ca2+ fluctuations, we currently think
are the major mechanisms for the antiarrhythmic effects of these PUFAs.
|