One of us (D.S.) will be releasing a book on experimental molecular modeling around 4-1-99 (Wuerz Publishing Ltd. , Winnipeg, Canada).
Below is a typical chemical analysis which can be found in the text:
Computer- Aided Analysis of a Possible Binding Conformation for d- Lysergic Acid Diethylamide (LSD) to the 5HT2A Receptor.
David S. Soriano and Robert J. Ellison, Department of Chemistry, University of Pittsburgh- Bradford. Bradford, Pa. 16701
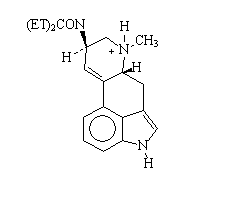
It has now been about 60 years since Hofmanns pioneering work with derivatives of lysergic acid and his accidental discovery of the pyschedelic agent d-lysergic acid N,N- diethylamide (1- LSD):
From studies relating structure and activity , it was noted more than 35 years ago that minor modifications in the ergot alkaloids produce surprising changes in pharmacological effect. (1).
Knowledge of the biologically-active conformations of lysergic acid derivatives would clarify understanding of the hallucinogenic properties
And of the nature of binding to the 5-HT2 receptor. (2)
LSD is known to dock inside the recognition site of the human serotonin 5-HT2A receptor , which is one member of the large G- protein coupled receptors. It is not known exactly how binding occurs to the recognition site, however, in all monamine neurotransmitter receptors there is an absolutely conserved aspartate residue that is believed to bind to the protonated N6 amine (3):
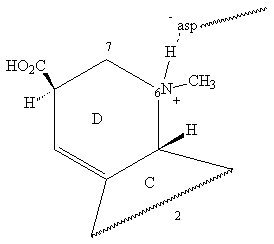
Although the lysergamides are relatively rigid , they have some conformational flexibility in the D ring and the carbonyl linkage (4)
The D ring being a cyclohexene derivative , can exist in a half-chair conformation with the N6 group "flap- up" or flap-down" (the C7 group will have an opposite relationship to the former):
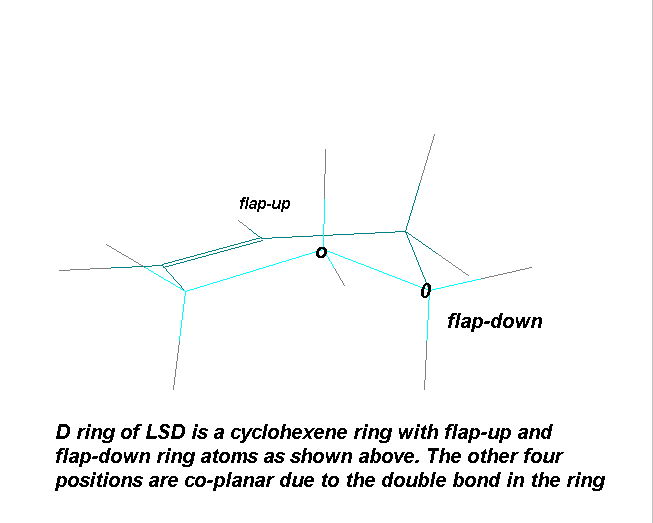
The Pierri group has reported that Ergotamine (the most typical ergolene from the ergot alkaloid group) exists in the C7 "flap-down" conformation in chloroform solution although this is still subect to debate (5)
From the pKb , it is certain that about 75% of the LSD is protonated at the N6 site. Whether this proton is abstrated by the asparate residue is not known.
The Nichols group has carried out molecular mechanics calculations on the
7C "flap-up" and "flap-down" conformational energies which has been corroborated by this author. It was clear from the calculations generated by the Nichols group that the protonated forms for the lysergamides studied
were more stable than the free base forms by 3-4 Kcal/ mole and the "flap-up" (C7) conformations were made stable than "flap-down" by 1-3 Kcal/ mole (6)
However, this author noted in the above paper that the possibility of intramolecular H bonding between the amide oxygen and the N6 proton
was excluded from the molecular mechanics studies. Since such an intramolecular attraction would require a "flap-up" conformation for the
6N (with psuedo equatorial placement of the N- methyl group) , it was decided to carry out such molecular mechanics studies in this report. It is quite possible that this conformation is retained even after docking is complete with the receptor.
Calculations were carried out with the "PC model" modeling program (Serena Software, Bloomington, In.) which uses the MMX force-field as developed by Gajewski and Gilbert. Calculations were carried out, in vacuo, with no effort to determine the effects of polar solvent on conformation.