Program For Genetics and Psychoses
Genetic Research Projects
It has been very challenging to find the genetic factors that predispose people to many severe complex diseases, including the psychotic disorders we are investigating. While we are still trying to find the genes that predispose people to complex disorders, we are also investigating other genetic factors that may be intimately tied to them. We are very interested in the mechanisms of thought processes, or cognition, since they can be severely impaired when someone falls ill. We are also investigating daily rhythms, also called circadian rhythms, that are frequently disrupted during periods of psychoses. Currently, we are conducting follow-up studies of Genome Wide Association Studies (GWAS) conducted by the Psychiatric Genomics Consortium. In particular, our goal is to build cell-based models to understand the role of Copy Number Variations (CNVs), specifically 15q11 and 22q11 deletions, as well as mitochondria function.
The overarching goal of our research is to find safe and effective treatment for severe psychiatric disorders. We believe that such treatment can be developed in a rational manner if we have a better understanding of the genetic causes and how they lead to illness.
Apart from the research on causative factors, we are separately evaluating novel treatment methods that may prove to be helpful in addition to conventional treatment.
We are dedicated to training the next generation of doctors, researchers, teachers, and therapists.
Genome Wide Association Studies (GWAS)
Associations of over 100 single nucleotide polymorphisms (SNPs) and schizophrenia (SZ) risk have been published recently. The basis for the associations is uncertain, though connections with known immune and auto-immune abnormalities in SZ have been proposed, particularly for SNPs in the human leukocyte antigen (HLA) region on chromosome 6p. Interest in the HLA associations has re-emerged following several recent GWAS publications. We hope to address the following questions:
The recent mega analyses have suggested multiple SNPs conferring risk for SZ. It is difficult to detect which SNPs affect risk, due to the substantial Linkage Disequilibrium (LD) in the HLA region, compounded by the modest risk due to individual SNPs.
Most of the significantly associated SNPs from the GWAS analyses do not cause obvious functional changes, so it is difficult to postulate a plausible model for SZ pathogenesis based on the current associations. Yet, other SZ research indicates three pathways to pathogenesis for the HLA associated SNPs: (i) inflammatory/infectious pathways, (ii) autoimmune abnormalities, (iii) non-immune related functions, such as migration and differentiation of neuronal cells. We are pursuing these leads.
Copy Number Variations (CNVs)
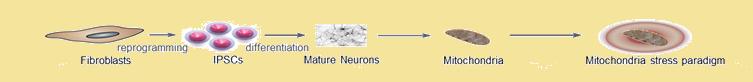
Copy number variations (CNVs)--the gain or loss of segments of genomic DNA relative to a reference, are associated with several complex and common disorders. CNVs provide a unique opportunity to understand pathogenic mechanisms of many human diseases. Currently, our lab is actively pursuing the 15q11 and 22q11 CNVs and their impact on dendritic spine density and mitochondrial function in neurons.
Chr22q11.2: A 1.5-3 Mb deletion on chromosome 22q11.2 occurs approximately once in 3-4000 births. Also called the DiGeorge or velocardiofacial syndrome (VCFS), it elevates risk for several neuropsychiatric disorders, as well as cardiovascular, craniofacial, limb, and immunologic abnormalities. Over 1/3 of deletion carriers are ultimately diagnosed with schizophrenia or schizoaffective disorder, and genome-wide association studies (GWAS) have repeatedly reported substantive associations with the deletion (odds ratios ~8).
Chr15q11.2: A deletion in the chromosome 15q11.2 deletion is a risk factor for neurodevelopmental delay, intellectual disability (ID), and neurodevelopmental disorders such as autism and schizophrenia. The deletion involves only four genes (all expressed in the brain) that are key components of processes such as magnesium transport (NIPA1 and NIPA2), microtubule cytoskeletal biogenesis and organization (TUBGCP5), and Cytoplasmic FMRP Interacting Protein (CYFIP1). CYFIP1 interacts with fragile X mental retardation protein (FMRP) and is a component of the WAVE Regulatory Complex, involved in dendritic spine generation and function. In rodents, under- or overexpression of CYFIP1 causes altered dendritic spine morphology, dynamics, and synaptic AMPA diffusion.
Mitochondrial studies
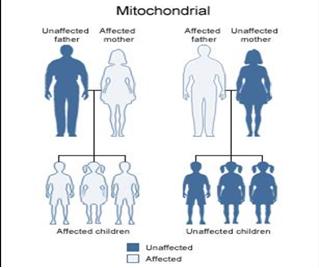
Mitochondria are the powerhouse of the cell and vital for oxidative phosphorylation. Unlike other organelles, mitochondria possess self-replicating, circular, and double-stranded DNA (mtDNA). The 16.6 kb circular mtDNA genome encodes 37 intronless genes. In humans, mtDNA is inherited almost exclusively from mothers and does not undergo recombination.
In individuals who have dysfunctional mitochondria, a common phenotype involves impaired brain function. There is suggestive evidence supporting the hypothesis that SZ is among the disorders resulting from mitochondrial failure, but the connection is far from clear. We are currently hoping to answer these questions:
SZ features are often present in patients with classical mtDNA diseases and can predate the typical neuromuscular features. Pedigrees segregating mtDNA diseases and SZ have also been reported. The psychiatric manifestations of mtDNA mutations include classical features of SZ, atypical psychoses, mood disorders, and cognitive deterioration with diffuse cortical atrophy.
Reduced mitochondrial number and abnormal mitochondrial morphology have been documented from muscle biopsies and in post-mortem brain tissue from SZ patients. Impairment in mitochondrial functional assays, mitochondrial dynamics, and protein levels has also been reported in SZ brain and peripheral tissues. Mitochondrial gene expression differences in post-mortem SZ case-control studies have been reported, but not replicated. These abnormalities could cause SZ or represent epiphenomena, such as medication effects.
Family studies show maternal inheritance for a proportion of SZ cases. Nominal associations between 'candidate' mtDNA variants and SZ risk have been reported, but there are inconsistencies. Only three studies have published a systematic screen for the entire mtDNA sequence. The differences were not observed between Bipolar cases and controls, suggesting specificity for SZ. Whether the mutations were inherited or occurred de novo is unknown.