This information is provided for those
who wish to use the differentiation of Naegleria gruberi for student
laboratories.
Differentiation of Naegleria and the
Isolation
and Characterization of Flagella
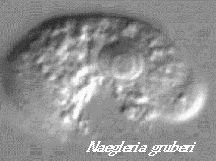
Introduction: Naegleria
gruberi is an amebo-flagellate. Amebo-flagellates are unusual organisms
that can exist as amebae
and also as swimming flagellates.
The ameboid stage feeds by phagocytosis of bacteria and divides by binary
fission. The flagellated stage does not feed or divide. Flagellates are
thought to provide a method of dispersal when food supplies become low.
Amebae are induced to become flagellates by removal
of their food supply, bacteria. This is done by gently washing the amebae
with ice-cold buffer in the centrifuge. The differentiation of amebae into
flagellates is started by suspending washed amebae in a warm-dilute buffer,
2 mM Tris-HCl, pH 7.6 at 25 oC in our experiments. This is defined
as 0 min. of the differentiation.
The differentiation of Naegleria provides a useful
system for examining a number of interesting problems in Cell and Developmental
Biology. Some of the advantages of the system stem from the ease and speed
with which cells can be grown and induced to differentiate. Another important
advantage is that the differentiation of amebae into flagellates is relatively
synchronous. This means that following the population of amebae as they
differentiate into flagellates is similar to following the changes in a
single cell. In this way we can ask about the molecular basis for the changes
that take place.
These changes include a disassembly of the actin-myosin
cytoskeleton that provides movement in amebae. This is reflected in the
first morphological change in the cells when they change from an ameboid
shape to a spherical shape at about 60 min. after initiation.
The next visible change is the appearance of short
flagella on the surface of the spherical cells. The time when 50% of the
population has developed visible flagella is defined as the T50 for flagella
formation. One of the great advantages of the Naegleria system is the reproducibility
of the differentiation. The T50 for flagellum formation (the
time for 50% of the cells to form visible flagella) is 68 +/- 2 min.
About the time flagella have reached three quarters
of their full-length, the cells change shape again. This time the spherical
cells become flattened ovals referred to as the flagellate shape. The T50
for flagellate shape formation is 85 min. Formation of the flagellate shape
is correlated with the formation of an extensive cytoskeleton composed
of microtubules. The T50 for cytoskeletal microtubule (CSMT) formation
is 75 min. This is particularly dramatic because amebae lack any kind of
microtubules except in the mitotic spindle.
Although not visible without staining for tubulin,
the de novo formation of basal bodies precedes the formation of flagella
by about 10 min. The de novo formation of basal bodies is a nearly unique
feature of the Naegleria differentiation and it provides the potential
for investigating the control of these poorly understood organelles.
Our lab today will involve the differentiation
of amebae into flagellates. We will follow this process by fixing cells
in Lugol's iodine and scoring for the presence of flagella. We will fix
cells in a mixture of formaldehyde and nonionic detergent for staining
of the cytoskeleton which will be done in two weeks. We will also collect
samples for the analysis of tubulin by Western blotting next week.
At the end of the differentiation we will harvest
flagellates and remove the flagella using a pH shock. We will prepare the
isolated flagella for examination by negative staining in the election
microscope in two weeks.
Materials:
-
plates of Naegleria gruberi grown on Klebseiella
pneumoniae. (see below).
-
ice-cold 2 mM Tris-HCl, pH 7.6, 1 liter.
-
Lugol's iodine (see below)
-
0.5% PMSF in n-propanol, 100X stock soln. (CAUTION
- TOXIC)
-
2 M leupeptin, 100X stock soln.
-
100 mM EGTA, pH adjusted to 8.25 with NaOH, = 100X
or 1000X stock.
-
100 mM dithiothreitol (DTT), = 1000X stock.
-
0.1 M Tris-HCl, pH 7.6, = 50X stock.
-
1 M MgCl2 stock.
-
100 mM sodium acetate, pH 3.7, prepared by adding
0.575 ml glacial acetic acid to about 85 ml DIW(deionized water) . The
pH is adjusted to 3.7 with 10 N NaOH and then the volume is brought to
100 ml with DIW. This is a 10X stock.
-
MgCF,
50 mM sodium phosphate, pH 7.2, 125 mM sucrose,
5 mM MgCl2, 1 mM EGTA, 0.1% w/v NP-40 (dilute from 10% w/v stock),
0.9% formaldehyde (dilute from 36% soln = formalin).
-
ice-cold Detailing Medium, 10 mM sodium acetate,
pH 3.7 [from 10X stock], 2 mM MgCl2, 75 mM sucrose, 0.1 mM EGTA
[from 1000X stock] and 0.005% PMSF and 20 mM leupeptin added just before
use from 100X stocks.
-
ice-cold Neutralizing Buffer, 0.5 M Tris-HCl, pH
8.25.
-
ice-cold Triton I Soln, 25 mM Tris-HCl, pH 7.6, 3
mM MgCl2, 1 mM EGTA, 0.1 mM DTT , 0.2% Triton X-100 (from a
10 w/v stock), 0.005% PMSF added just before use.
Procedures:
A) Differentiation of Amebae.
-
1) Put 20 ml of 2 mM Tris-HCl, pH 7.6 (Tris) into
a 125 ml Erlenmeyer flask and put the flask in a reciprocating water bath
set at 25.0 oC. Allow the buffer to reach the bath temperature
before starting to harvest cells. Meanwhile collect an ice bucket with
a 50 ml polycarbonate centrifuge tube, a similar tube for use as a balance,
and ice-cold Tris with a 10 ml pipette.
IMPORTANT, set up the tubes in Part B,
steps (1), (2) and (3) before proceeding with the differentiation.
-
2) Suspend each plate of amebae in 9 ml ice-cold
Tris using a glass spreader. DO NOT push down on the spreader, it will
break. Pour the cell suspension into a 50 ml polycarbonate centrifuge tube
in ice. One tube will hold the cells from 5 plates.
-
3) Prepare a balance tube with an equal volume of
water then wash the amebae free of bacteria by centrifugation at a setting
of 6 in the Clinical centrifuge for 45 sec. Bring the head to a stop rapidly
using the brake in the cover. DUMP the supernatant from the tube, place
the tube in ice, and rapidly resuspend the cells by pipetting in 10 ml
of ice-cold Tris. NOTE: In contrast to most cases, the supernatant is not
carefully decanted because this results in a loss of the cells in the loose
pellet. DUMP means to rapidly invert the tube and then immediately return
it to an upright position. This may not remove all of the supernatant but
that is not a concern. Your instructor will demonstrate.
-
4) Vortex the cell suspension to remove bacteria
adhering to the amebae. Repeat the centrifugation, DUMP, and resuspension
as in (2). Note: The intention is to minimize the time amebae are in a
pellet and the time from initial resuspension until the cells are put into
warm buffer.
-
5) Repeat the centrifugation and DUMP as in (2) but
this time resuspend the cells in 10 ml of 25 oC Tris. START
a timer, this is zero time for the differentiation. Pour the cell suspension
into the 125 ml Erlenmeyer flask with the remaining 10 ml of warm Tris
and place the flask on a reciprocating shaker water bath at 25 oC
and 80-100, 1 inch strokes per min.
B) Following the Differentiation.
-
1) Prepare a set of 16, 10 x 75 mm or 13 x 100 mm
test tubes at room temperature with one drop of Lugol's iodine in each.
When adding the iodine soln it is IMPORTANT to hold the dropper
well away from the lip of the tube and try and get the drop to fall to
the bottom of the tube without touching the walls. Any iodine that is present
on the lip of the tube may be carried back to the cell suspension and this
would kill all the cells. Label the tubes 0, 10 ,20, 30, 40, 50, 60, 65,
70, 75, 80, 85, 90, 100, 110, and 120 min.
-
2) Prepare three 10 x 75 mm disposable test tubes
in
ice with 250 µl of MgCF in each. Use care to place the fixative
in the bottom of the tube so as not to contaminate the lip (see above).
Label these tubes 10, 70, and 120 min.
-
3) Prepare a set of 6, 1.5 ml microcentrifuge tubes
with boiling resistant labels at room temperature. Label the tubes 5, 20,
40, and 80 min.
-
4) Using a wide bore sampling pipette to prevent
shear, take a sample from the flask of cells and add 3 drops to a tube
containing Lugol's iodine at the times indicated. Try and add the cells
so that they fall directly into the iodine soln rather than down the side
of the tube but take special CARE not to touch the sampling pipette
to the tubes containing Lugol's iodine. Gently shake each tube after
addition of the cells. Note that the "0" time point will be a little late.
Record this time in your notebook.
IMPORTANT: DO NOT return unused
cells to the flask. Discard any cells remaining in the sampling pipette
into the DUMP. After each sample, rinse the sampling pipette three times
in DDW and store in DDW.
-
5) At 5, 20, 40, and 80 min. after the initiation
of the differentiation remove 1 ml of the cell suspension to a microcentrifuge
tube with a P1000. Centrifuge for 30 sec. and carefully aspirate the supernatant.
Immediately place the tube with the cell pellet at -20 °C. We will
run the gels next week.
-
6) At 10, 70, and 120 min. also add a sample of four
drops of cell suspension to a tube containing MgCF using the same CARE
as above to avoid contaminating the flask of cells.
C) Isolating Flagella.
Note: Before starting this section make sure you
understand the steps and that you have all the reagents and pipettes on
hand. It is IMPORTANT that steps (1) and (2) be carried out quickly.
-
1) After you have taken samples to Lugol's iodine
and MgCF at 120 min., pour the cells remaining in the flask into a 50 ml
screw cap centrifuge tube (you do not need to put the cap on at this step)
and centrifuge at a setting of 6 for 2 min. in the Clinical centrifuge.
Decant the supernatant and place the tube in ice.
-
2) Remove the flagella by adding 10 ml of ice-cold
Detailing medium, cap the tube, and shake vigorously for 15 sec. IMMEDIATELY
add 0.5 ml of Neutralizing buffer and mix.
-
3) Remove the cell bodies by centrifuging at a setting
of 6 for 2 min. in the Clinical centrifuge.
-
4) Pour the supernatant into an ice-cold 15 ml centrifuge
tube and centrifuge at 600 x g for 1 min. in the cold to remove residual
cell bodies and food vacuoles.
-
5) Pour the supernatant into a second ice-cold 15
ml centrifuge tube and centrifuge at 600 x g for 4 min. in the cold.
-
6) Pour the supernatant into a third ice-cold 15
ml centrifuge tube. Remove a small drop of the flagellar suspension to
a slide and examine it as described under (D). Record your observations.
Centrifuge the suspension at 20,000 x g for 10 min. in the cold to pellet
the flagella.
-
7) Discard the final supernatant and resuspend the
flagella, using a Pasteur pipette, in 5 ml of ice-cold Triton I solution
to remove the flagellar membrane from the flagellar axoneme. Transfer this
suspension to an ice-cold 12 ml Corex centrifuge tube. Incubate on ice
for 15 min.
-
8) Pellet the axonemes by centrifugation at 40,000
x g for 30 min. in the cold.
-
9) Discard the supernatant and resuspend the pellet
in 250 µl of ice-cold Triton I solution using a Pasteur pipette.
D) Making Slides for Staining the
Cytoskeleton.
-
1) Using a diamond pencil, label the end of two slides
for each of the three time points with the time and your group number.
-
2) Lay the slides flat under the hood with the labeled
side up. Gently shake the cells fixed in MgCF to suspend the cells and
then place 60 µl of the cell suspension as a puddle in the middle
of the slide. Allow to dry thoroughly under the hood.
-
3) Rinse the slides of each time point briefly
in PBS, drain for a min. and then transfer to MeOH in ice for 10 min. After
the MeOH fixation drain the slides briefly and then fix in acetone
in ice for 10 min. At the end of the acetone fixation, allow the slides
to dry under the hood at room temperature.
-
4) Store the dry slides at -20 oC over
desiccant.
-
To stain the slides, warm them to room temperature,
place a drop of anti-tubulin on the slide and spread gently
with the tip of a pipette. Incubate in a damp box at 37 oC for
60 min, wash by passing through 4 changes of TBS or PBS for 2-3 min. each.
Add second antibody (Alexa488 from Molecular probes is very good at 1/200)
in the same way and incubate . After washing, mount slides in 90% glycerol,
10% 0.1 M NaCO3, pH 9.0 containing 100 mg/ml of 1,4 - diazabicyclo-[2.2.2]
octane (DABCO) (3).
E) Preparing EM Grids of Flagellar
Axonemes.
1) Pick up an EM grid, coated with Parlodian and
carbon, in the tips of a fine locking forceps. Place a small drop of the
axoneme suspension on the grid and allow to stand for 30 sec. Remove the
drop by touching the edge of a piece of filter paper to the edge of the
grid and allowing the drop to be soaked into the paper. Immediately place
a drop of 1% uranyl acetate on the grid allow to stand for about 15 sec.
Remove the uranyl acetate drop with the edge of a piece of filter paper.
Allow the grid to air dry.
G) Counting Flagellates. (Probably
save for later. Cover the tubes tightly with Parafilm).
-
1) Prepare a counting slide by placing three 18x18
mm cover slips supported by small clay feet on the slide. Resuspend the
cells fixed in Lugol's iodine by gently shaking the tube and then place
a drop of fixed cells under a cover slip. Repeat for two other samples.
You may find it easier to start counting with the 120 min. sample and working
backward.
-
2) Examine the cells using a 40X phase contrast objective.
Count the fraction of cells with one or more flagella in fields chosen
at random. Count only cells that are completely within the field and that
are not touching another cell. It is IMPORTANT that you focus up
and down on each cell as you examine it to see if it has flagella. The
flagella are so thin that they can be out of the plane of focus when you
can see the cell body. If you are unsure whether a short projection is
a flagellum, do not count it. Count cells from fields chosen at random
until you have scored 100 cells at each time point. Record the time of
the sample and the percent flagellates. When you have all the data, plot
the percent flagellates vs. the time of sampling.
H) SDS Gels and Western Blots.
(Week 2).
-
1) Run the samples on on a
7% SDS gel.
-
2) Blot the gel to nitrocellulose
or PVDF
-
3) Develop the blot with an
anti-tubulin first antibody and a HRP or alkaline phosphatse second antibody
and appropriate substrate.
I) Growing Naegleria. (see references
4 and 5 for more details)
-
1) Pour NM plates:
-
to 800 ml of DIW add:
-
1.2 gm K2HPO4
-
0.8 gm KH2PO4
-
1.6 gm dextrose
-
1.6 gm Bacto-peptone
-
after these are completely
dissolved add
-
16 gm Bacto-agar without
mixing
-
autoclave for 15 min.
-
mix well by swirling soon after
removal from the autoclave but after bubbling has stopped.
-
cool to about 50 oC
and
pour about 40 ml per plate
-
dry plates at room temperature
for 3 days before use and then store at 3 oC
-
2) Prepare Penassay broth:
-
to 80 ml of DIW add:
-
1.4 gm of Difco Antibiotic
medium #3 and dissolve
-
aliquot 8 ml into 16 x 150
mm culture tubes, cap and autoclave for 15 min.
-
store at room temperature.
-
3) Inoculate Klebsiella
pneumoniae from a slant into Pennassay broth and incubate at 34 oC
for
24 to 48 hr. This stock is good for 1 week stored at room temperature.
Inoculate from a slant to broth every week do not inoculate from broth
to broth as the cultures become slimy (5).
-
4) Prepare a Naegleria edge
plate.
-
Place 0.1 ml of a K. pneumoniae
broth
culture on an NM plate and spread with a glass spreader.
-
Using a sterile inoculating
loop place a small dot of Naegleria cysts near one edge of the plate.
-
Incubate the plate inverted
at 34 oC for 2-3 days during which
an "edge" of growing amebae will move across the plate as they consume
the bacteria.
-
When the edge has reached the
far side, the plate will be covered with cysts. Wrap the plate tightly
in Saranwrap ( You must use the thick plastic.) and store at room
temperature for 1-3 weeks.
-
5) Prepare spread plates:
-
A few hours before inoculating,
place the NM plates at room temperature to warm up.
-
22 hr. before use:
-
Use a sterile inoculating loop
to scrape up cysts from an edge plate.
-
Suspend the cysts in sterile
DIW and vortex vigorously to disperse.
-
Count the cysts with a haemocytometer
and adjust the concentration to 2.5x106 cysts/ml.
-
Place 0.1 ml of K. pneumoniae
broth
culture on each NM plate.
-
Place 0.1 ml of the cyst suspension
(vortex just before use as the cysts settle rapidly) on each plate.
-
Using a sterile glass spreader,
mix and distribute the bacteria and cysts evenly over the plates.
-
Incubate the plates, right
side up, at 34 oC for 1-2 hr and
then invert the plates.
J) Preparing Lugol's Iodine (5).
-
Using a glass bottle
with a ground glass stopper:
-
add 2 gm of iodine to the bottle
(CAUTION - TOXIC)
-
add 3 gm of potassium iodide
to the bottle
-
add 0.5 - 1.0 ml of DIW
( may need a little more)
-
mix until dissolved
-
add DIW to a total of 50 ml
-
store at room temperature
References:
-
1) Fulton, C. and A.D. Dingle. 1967. The appearance
of the flagellate phenotype in populations of Naegleria. Develop. Biol.
15:165-191.
-
2) Dingle, A.D. and C. Fulton. 1966. Development
of the flagellar apparatus of Naegleria. J. Cell Biol. 31:43-54.
-
3) Walsh, C. 1984. Synthesis and assembly of the
cytoskeleton of Naegleria gruberi flagellates. J. Cell Biol. 98:449-456.
-
4) Fulton, C. 1993. Naegleria: A research partner
for cell and developmental biology. J. Euk. Microbiol. 40:520-532.
-
5) Fulton, C. 1970. Amebo-flagellates as research
partners: The laboratory biology of Naegleria and Tetramitus. Meth. Cell
Physiol. 4: 341-476.
{kind=link}
{kind=link}